One of the major issues encountered during drug discovery and development is compound’s low aqueous solubility resulting in low bioavailability and developability issues. Various approaches are employed for increasing the bioavailability of poorly water-soluble drugs that include salt formation, solubilisation using a co-solvent, complexation with cyclodextrin and particle size reduction; all these approaches have various limitations. Amorphous solid dispersion is one of the most promising approaches to enhance bioavailability and to enable compounds to toxicity studies, overcoming the drawbacks of the other approaches. Solid dispersion approach has unique advantages in terms of its applicability for wide range of molecules, safety of vehicles and scalability.
Solid dispersion refers to the dispersion of one or more active ingredients in a hydrophilic inert carrier matrix at molecular level. It is prepared by the melt (fusion) method and solvent evaporation technique. The basic principle of this technique involves conversion of crystalline drug substance to amorphous form and stabilised in a hydrophilic polymer. When the solid dispersion comes in contact with the aqueous medium, the inert carrier dissolves and the drug is released, the increased surface area produces a higher dissolution rate thereby increasing the bioavailability of the poorly soluble drug substance.
In this case study, we would explain how Aragen’s formulation scientists devised a formulation strategy and protocol to develop a robust solid dispersion formulation for a poorly soluble compound to achieve the desired bioavailability and in vivo exposure.
The client is a biotech company based in Europe and focused on drug discovery and development of new chemical entities (NCE). The project goal was to develop orally bioavailable amorphous solid dispersion formulation for a poorly soluble compound using Aragen’s proprietary protocol and technology. The compound is a crystalline material with a melting point of 203o C and solubility in FaSSIF of 2µg/mL and oral bioavailability less than 3%.
Aragen devised a systematic screening and development approach, over a period of 4 weeks, to identify suitable inert carrier i.e. hydrophilic polymer and surfactant to obtain a stable, amorphous solid dispersion using spray drying process.

Step 1: Polymer Screening: (12 trials – 10% drug load, 20mg per trial, Duration – 1 week)
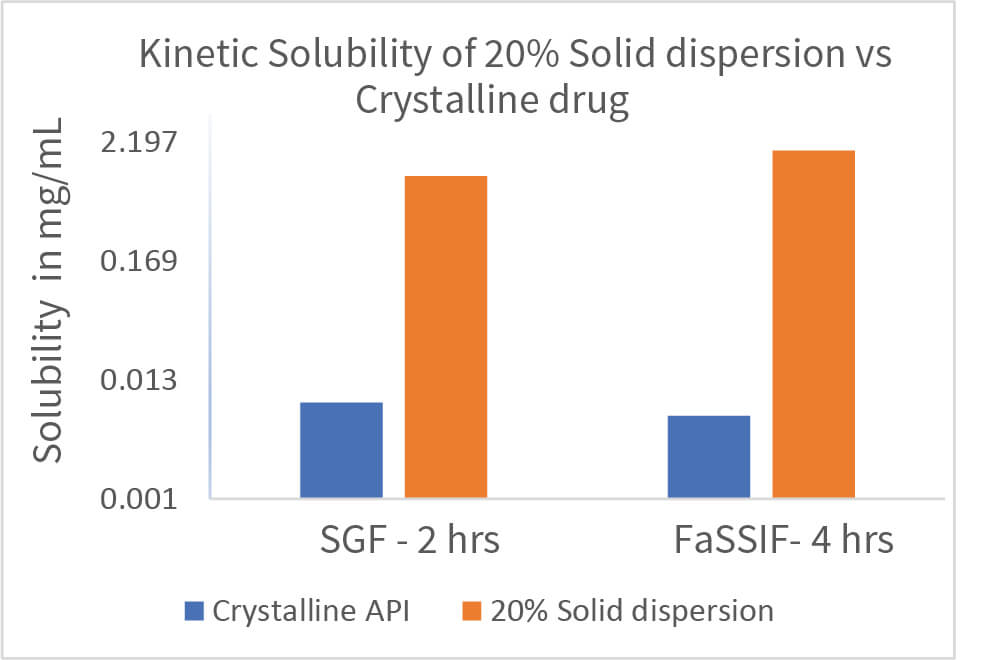
The solid dispersion screening was conducted by freeze drying process. Appropriate solvent system for freeze drying process was selected by solubility screening of drug substance in organic solvents. The screening was designed with 12 polymers and 20mg drug substance and 180mg polymer. Drug and polymer were dissolved in the solvent and subjected to freeze drying cycles using a Lyophiliser to obtain a free-flowing powder. The resulting solid dispersion powders were characterised for PXRD, mDSC (Tg), Assay, Purity and Kinetic solubility in SGF and FaSSIF. The best polymers were selected for the next stage based on the characterization data.

Step 2: Polymer & Surfactant Screening: (15-20 trials – 20mg per trial, Duration – 1 week)
The screening involved 2-3 best polymers selected from the step-1 screening and 8 surfactants with 20mg compound per trial. The screening was conducted using the same process outlined in step-1 screening. The best polymer and surfactant combinations were selected for the next stage based on the characterization data.
Step 3: Drug load optimization: (5-10 trials – 50mg per trial, Duration – 1 week)
The screening involved 2-3 best formulations selected from step-2 screening and evaluating drug loading upto 50%w/w using same process outlined in step-1 screening. The best formulations were selected for scaleup and stability studies based on the characterization data.
Step 4: Scale up and Stability studies: (2-3 trials – 500mg per trial, Duration – 2 weeks)
Scale up batches were prepared for selected formulations and loaded for stability studies at 2-8o C, 25o C/60%RH, 40o C/75%RH for 4 weeks. The batches were characterized for PXRD, mDSC, TGA, DVS, SEM, Kinetic solubility, Assay and Purity. One formulation was selected for in vivo PK study based on the 1-week stability data followed by execution of PK study in rat.
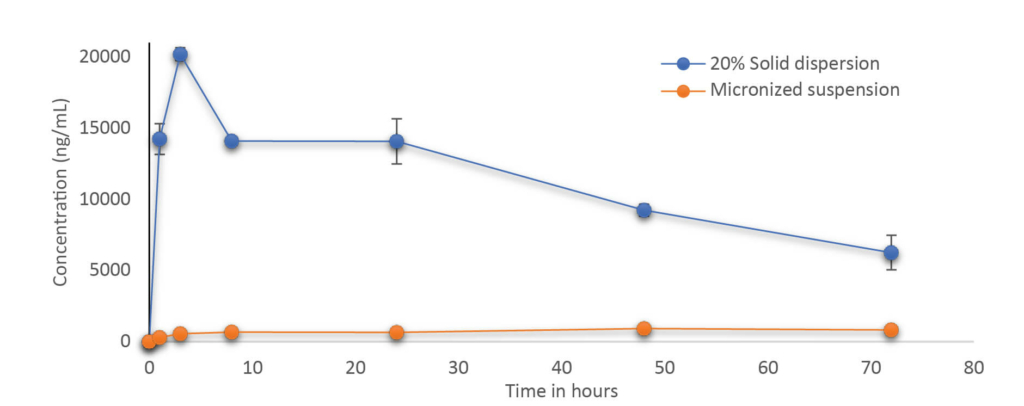
A stable solid dispersion was successfully developed at 20% drug loading. The solid dispersion formulation was physically and chemically stable for 4 weeks. In vivo study demonstrated that solid dispersion significantly improved the bioavailability & plasma concentration compared to microsuspension by several folds paving the way for further development of the molecule with a clear formulation strategy to support Toxicity and Clinical studies.